Our research interests include: Please click on tabs for more details
- 1. Allosteric site discovery, ranking and characterization
- 2. Novel approach to enzyme engineering
- 3. Role of cyclophilins in retrovirial infection
- 4. Characterizing biomolecular dynamics with neutron scattering
- 5. Molecular dynamics on high-performance computing and heterogeneous architectures (including GPUs)
- 6. Open Source Software (OSS) for scientific computations
Allosteric Site: Discovery and Characterization
Allosteric modulation of protein presents unique opportunities for discovery of new medicine with novel mechanism of target control. In addition to inhibition (negative modulation), allostery provides opportunities of positive allosteric modulation. We have recently developed a unique computational methodology that allows identification, ranking and characterization of allosteric sites on protein targets of interest. This technology is based on identification of conserved network pathways of interactions between the protein surface and active-sites, and provides biophysical insights into the mechanism of allosteric communication.
Our unique approach is based on the biophysical mechanism of allosteric communications in enzymes. See the enzyme sub-section (see the tabs above) for more details or contact us for more details.
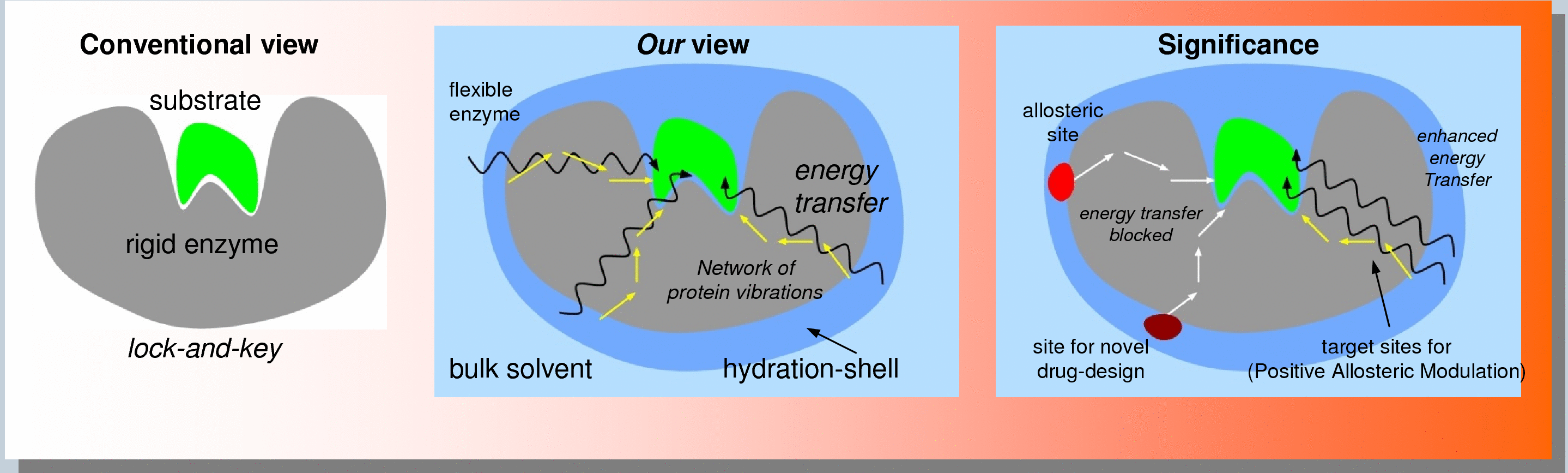
- The unique advantages of our approach are:
- Speed: We identify the allosteric site through computational modeling. We characterize and rank them by impact on the target activity.
- Low Cost: High throughput screening are expensive and do not provide the location of ligand binding. We provide a list of allosteric sites and a list of potential ligands that bind on the allosteric sites.
- True allosteric sites: Not all sites where ligands binds will control target activity, our approach only focuses on the true allosteric sites that change (negatively or positively) the activity.
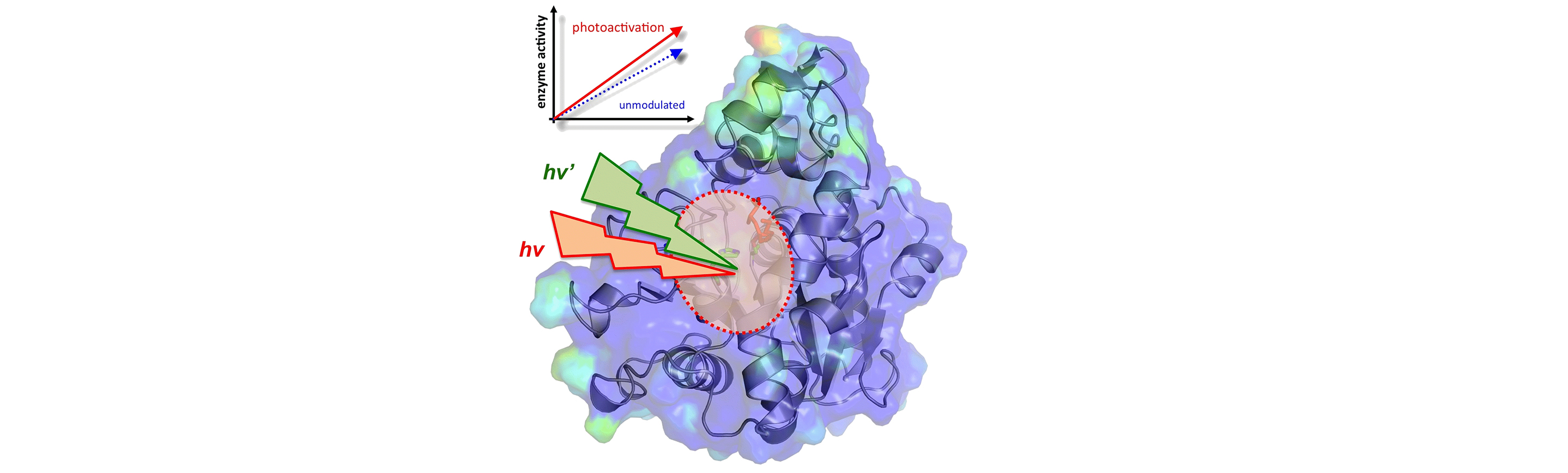
Enzyme engineering: Hyper-catalytic enzymes through conformational modulation
Enzymes undergo constant conformational fluctuations. Our ongoing work is revealing that a number of conformational fluctuations are conserved part of enzyme structure and they play an important role in enzyme rate-enhancement. See below for more details on our computational modeling of enzyme conformational fluctuations and enzyme dynamics in relation to promoting the designated function.
We are developing hyper-catalytic enzymes through identification of the reaction promoting conformational fluctuations, and modulating them through external signal such as light of appropriate wavelengths. We have used a novel chemical modification of enzyme Candida antarctica lipase B (CALB) that allows modulation of the enzyme conformation to promote catalysis. Computational modeling was used to identify dynamical enzyme regions that impact the catalytic mechanism. Surface loop regions located distal to active site but showing dynamical coupling to the reaction were connected by a chemical bridge between Lys136 and Pro192, containing a derivative of azo-benzene. The conformational modulation of the enzyme was achieved using two sources of light that alternated the azo-benzene moiety in cis and trans conformations. Computational model predicted that mechanical energy from the conformational fluctuations facilitate the reaction in the active-site. The results were consistent with predictions as the activity of the engineered enzyme was found to be enhanced with photo-activation. Preliminary estimations indicate that the engineered enzyme achieved 8 to 52 fold better catalytic activity than the unmodulated enzyme. See this article for more details.
Understanding the interconnection between enzyme structure-dynamics-function
Enzymes are highly efficient catalysts. A number of theories have been proposed to explain enzyme catalysis; however, detailed understanding of the biophysical mechanism of enzymes and the factors that contribute to catalytic efficiency remains limited. Over the last century, protein structure has been emphasized as the key to understanding protein function including enzyme catalysis. Increasing evidence from experimental and computational investigations, however, continues to reveal that proteins are not rigid structures but constantly undergoing conformational fluctuations.
An integrated view of protein structure, flexibility and function is emerging, where proteins are considered intrinsically flexible molecules; and the internal motions of a protein are closely linked to its designated function such as enzyme catalysis. In this new paradigm the hydration-shell and bulk solvent thermodynamical fluctuations also have an important affect on the protein motions and, therefore, protein function. Our long term goal is to understand interrelation between the enzyme structure, flexibility and function. At present, we are investigating the role of protein motions and conformational fluctuations in enzyme mechanism and the rate-enhancement achieved by enzymes. Our preliminary computational investigations of medically important enzyme cyclophilin A (CypA) has lead to discovery of a network of protein motions, which plays a role in promoting the catalytic step. The discovered network spans from the flexible surface loop regions all the way to the active-site, and is conserved as a part of the protein structure. Further, investigations of hydride transfer catalyzed by a plasmid encoded R67 dihydrofolate reductase (R67 DHFR) have provided valuable insights into the link between increased motions and enzyme kinetics. Currently, we are also investigating hydride transfer in dinucleotide binding Rossmann fold proteins (DBRPs) super-family. Based on early evidence, we hypothesize that:
- 1. Conformational fluctuations and protein motions play a promoting role in enzyme mechanisms
- 2. Protein motions enable energetic coupling between the solvent and the enzyme surface residues
- 3. The energy from solvent is transferred to the active-site through a network of residues
- 4. In the active-site the protein motions and the energy from solvent promotes the reaction by facilitating the attainment of the transition state.
We are investigating various aspects of enzymes to understand the factors that enable the high catalytic efficiency.
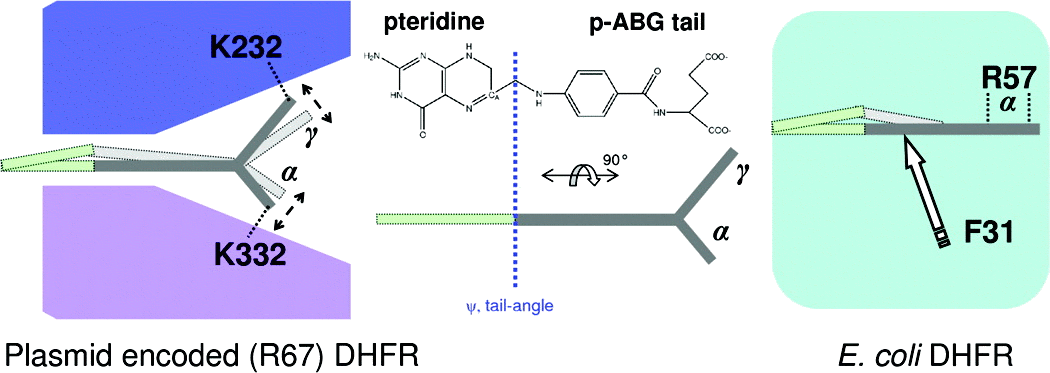
Comparing Non-homologous Enzymes Catalyzing Same Chemical Reaction
Plasmid-encoded R67 dihydrofolate reductase (DHFR) catalyzes a hydride transfer reaction between substrate dihydrofolate (DHF) and its cofactor, nicotinamide adenine dinucleotide phosphate (NADPH). R67 DHFR is a homotetramer that exhibits numerous characteristics of a primitive enzyme, including promiscuity in binding of substrate and cofactor, formation of nonproductive complexes, and the absence of a conserved acid in its active site. Furthermore, R67’s active site is a pore, which is mostly accessible by bulk solvent. This study uses a computational approach to characterize the mechanism of hydride transfer. Not surprisingly, NADPH remains fixed in one-half of the active site pore using numerous interactions with R67. Also, stacking between the nicotinamide ring of the cofactor and the pteridine ring of the substrate, DHF, at the hourglass center of the pore, holds the reactants in place. However, large movements of the p-aminobenzoylglutamate tail of DHF occur in the other half of the pore because of ion pair switching between symmetry-related K32 residues from two subunits. This computational result is supported by experimental results that the loss of these ion pair interactions (located >13 Å from the center of the pore) by addition of salt or in asymmetric K32M mutants leads to altered enzyme kinetics [Hicks, S. N., et al. (2003) Biochemistry 42, 10569−10578; Hicks, S. N., et al. (2004) J. Biol. Chem. 279, 46995−47002]. The tail movement at the edge of the active site, coupled with the fixed position of the pteridine ring in the center of the pore, leads to puckering of the pteridine ring and promotes formation of the transition state. Flexibility coupled to R67 function is unusual as it contrasts with the paradigm that enzymes use increased rigidity to facilitate attainment of their transition states. A comparison with chromosomal DHFR indicates a number of similarities, including puckering of the nicotinamide ring and changes in the DHF tail angle, accomplished by different elements of the dissimilar protein folds.
Discovering Conformational Sub-States Relevant to Protein Function
Internal motions enable proteins to explore a range of conformations, even in the vicinity of native state. The role of conformational fluctuations in the designated function of a protein is widely debated. Emerging evidence suggests that sub-groups within the range of conformations (or sub-states) contain properties that may be functionally relevant. However, low populations in these sub-states and the transient nature of conformational transitions between these sub-states present significant challenges for their identification and characterization.
To overcome these challenges, with Dr. Chennubhotla lab (University of Pittsburgh), we developed a new computational technique, quasi-anharmonic analysis (QAA). QAA utilizes higher-order statistics of protein motions to identify sub-states in the conformational landscape. Further, the focus on anharmonicity allows identification of conformational fluctuations that enable transitions between sub-states. QAA applied to equilibrium simulations of human ubiquitin and T4 lysozyme reveals functionally relevant sub-states and protein motions involved in molecular recognition. In combination with a reaction pathway sampling method, QAA characterizes conformational sub-states associated with cis/trans peptidyl-prolyl isomerization catalyzed by the enzyme cyclophilin A. In these three proteins, QAA allows identification of conformational sub-states, with critical structural and dynamical features relevant to protein function. Overall, QAA provides a novel framework to intuitively understand the biophysical basis of conformational diversity and its relevance to protein function.
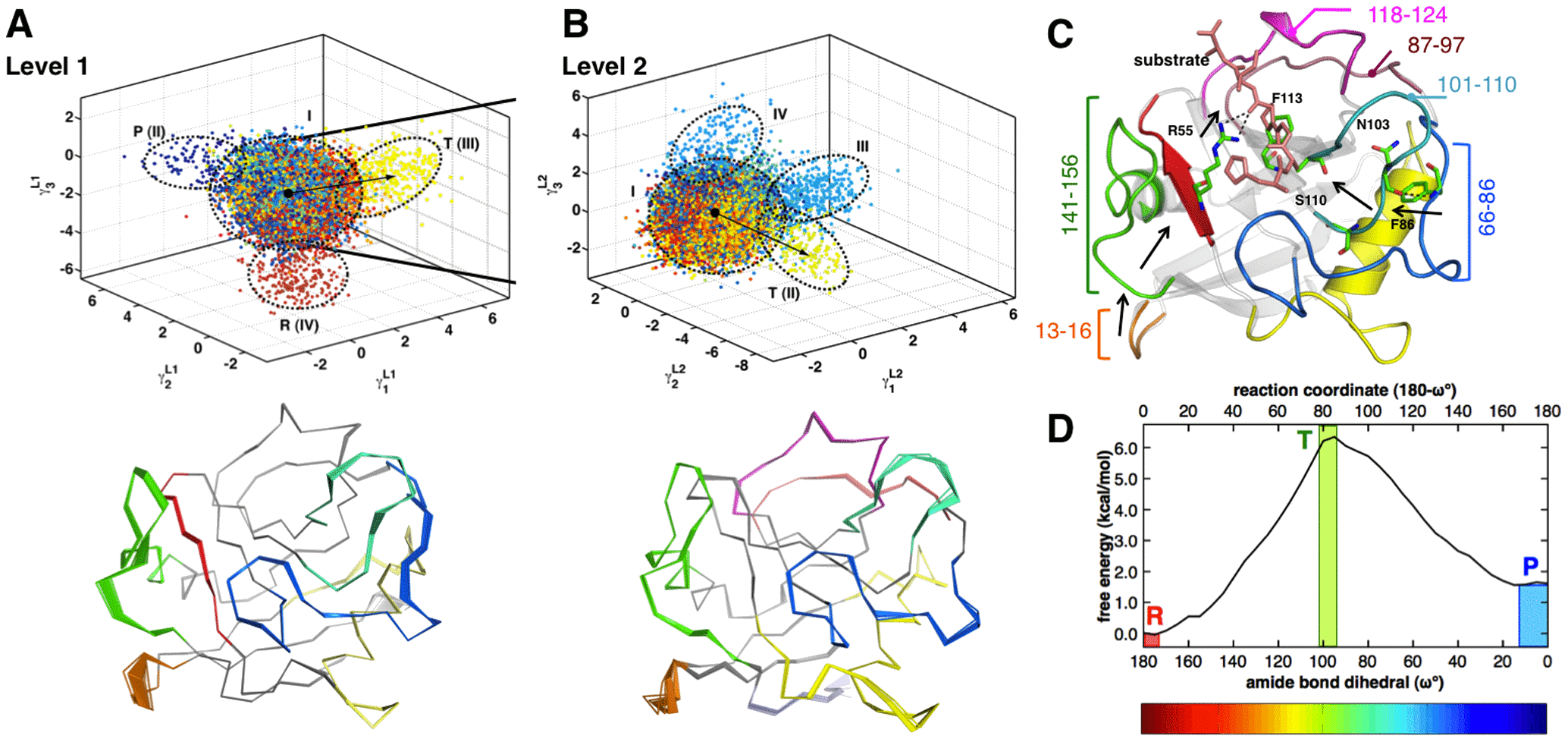
Figure: QAA describes conformational sub-states leading to transition state during catalysis in cyclophilin A.
Role of Cyclophilins in Retrovirial Infection
Cyclophilins are a family of proteins that bind to the immuno-suppresent drug cyclosporin. Members of this family have peptidyl-prolyl isomerase (PPIase) activity as they catalyze the isomerization of the peptide bond preceding proline residue in peptide and proteins. Cyclophilins have been known to play role in many cellular function including protein folding.
We are particularly interested in the human cyclophilin A (CypA) and cyclophilin B (CypB) due to their role in retrovirial infections. CypA is required for the infectious activity of Human Immuno-deficiency Virus type 1 (HIV-1) and CypB is known to promote the infectious activity of Hepatitis C Virus (HCV). The exact mechanism of the role of CypA in HIV-1 remains a topic of debate. However, it is known that CypA is incorporated into the virion in complex with Gag-encoded capsid protein. Some interest in the PPIase activity in relation to HIV-1 has also been expressed but investigations have revealed that PPIase activity may not be essential for the infectious activity. For CypB recent evidence suggests that interaction with non-structural protein 5B (NS5B) of HCV may promote the RNA dependent RNA polymerase activity.
We have developed computational models for investigating the PPIase activity of CypA in peptides and HIV-1 capsid protein. We have used a series of umbrella sampling type simulations to model the entire reaction pathway of cis/trans isomerization catalyzed by CypA. The details of the catalytic mechanism have revealed the role of various protein residues in the isomerization step. Further the computational models have also revealed details of protein-protein interactions between CypA and HIV-1 capsid protein.
The PPIase activity of CypA has also been used as a prototypical system for investigating the interconnection between enzyme structure, dynamics and function. We have discovered a network of residues in enzyme CypA that ranges from the flexible surface loops of the enzyme and reaches all the way into the active-site. The motions of these network residues are play a promoting role in the PPIase mechanism. Moreover, it has been discovered that these rate promoting motions (or slow conformational fluctuations) are conserved feature of the CypA fold (click on the movie below).
For CypB, to understand the possible role that this molecule plays in catalytic activity of NS5B we have prepared a computational model of the CypB-NS5B complex. This has been particularly challenging because no crystal structure of the complex is available. We are particularly interested in the hypothesis that the PPIase activity of CypB may alter the conformation of the NS5B tail to open up the active-site for RNA polymerase activity and therefore promoting the HCV infectious activity
Characterizing biomolecular dynamics with neutron scattering
Proteins are dynamic objects, constantly undergoing conformational fluctuations, yet the linkage between internal protein motion and function is widely debated. We are using joint computational-neutron scattering investigations to obtain insights into the role of protein motions in protein function.
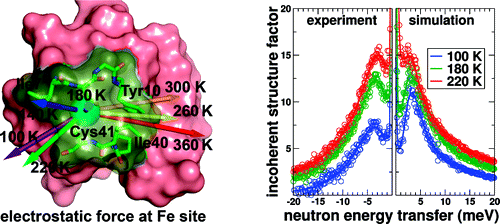
Recently, we have used this strategy to characterize temperature-activated collective and individual atomic motions of oxidized rubredoxin, a small 53 residue protein from thermophilic Pyrococcus furiosus (RdPf). Computational modeling allows detailed investigations of protein motions as a function of temperature, and neutron scattering experiments are used to compare to computational results. Just above the dynamical transition temperature which marks the onset of significant anharmonic motions of the protein, the computational simulations show both a significant reorientation of the average electrostatic force experienced by the coordinated Fe3+ ion and a dramatic rise in its strength. At higher temperatures, additional anharmonic modes become activated and dominate the electrostatic fluctuations experienced by the ion. At 360 K, close to the optimal growth temperature of P. furiosus, simulations show that three anharmonic modes including motions of two conserved residues located at the protein active site (Ile7 and Ile40) give rise to the majority of the electrostatic fluctuations experienced by the Fe3+ ion. The motions of these residues undergo displacements which may facilitate solvent access to the ion.
Molecular dynamics on heterogeneous architectures (including GPUs)
Molecular simulations using high performance computing (HPC) continue to play an important role in many domains including biology, chemistry and material science research. The future HPC architectures, on the way to and at Exascale, will pose significant performance challenges to applications including molecular simulations.
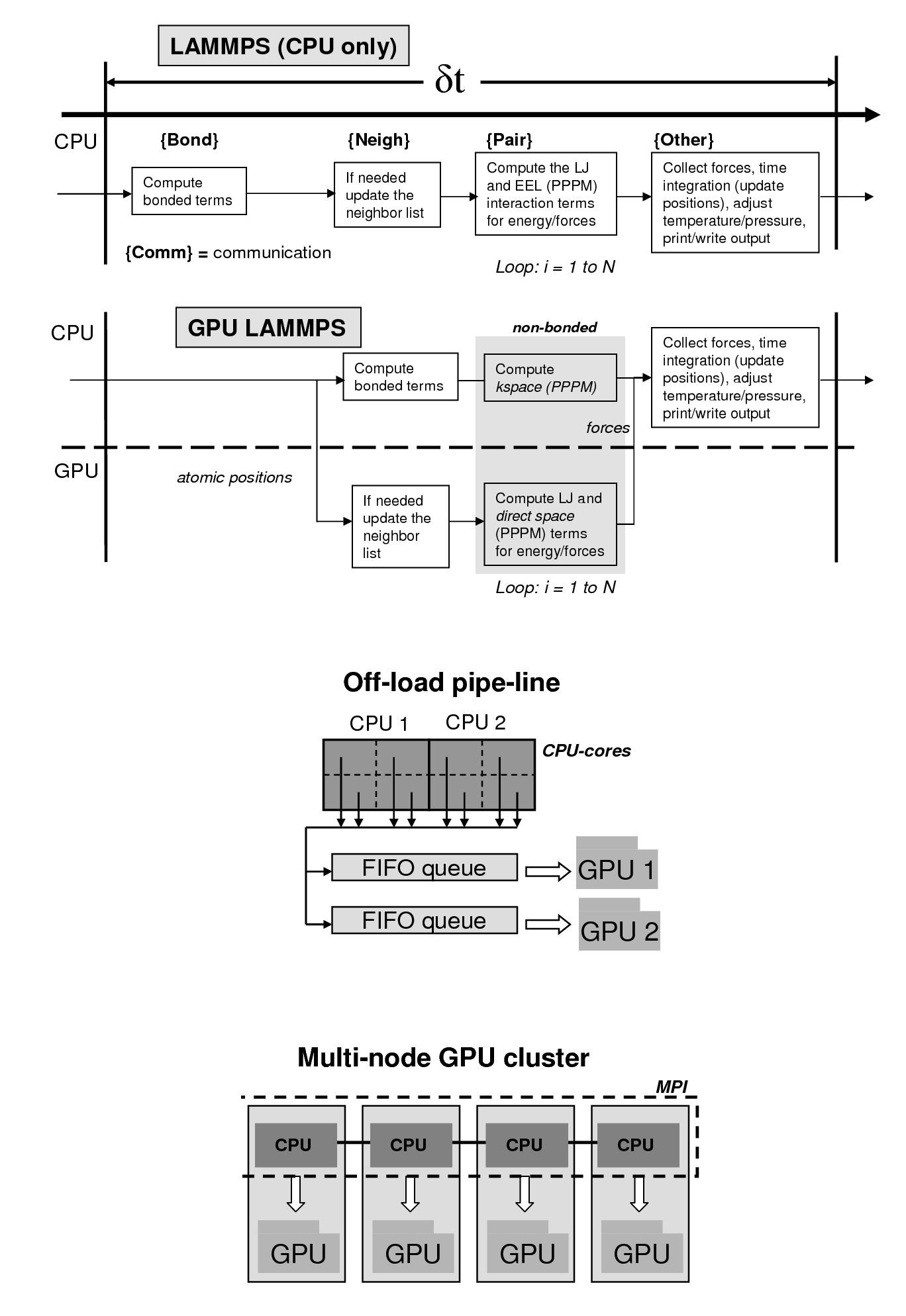
These future architectures are expected to deviate from the conventional path of concurrency in a homogeneous environment. The future systems are expected to have up to a billion way concurrency based on a multi-level hierarchy of heterogeneous resources. The heterogeneity will come from use of multi-core processors coupled with accelerators such as graphics processing units (GPUs). In order to harness the potential of these emerging architectures, codes and algorithms will need to optimally utilize the hardware resources. We hypothesize that a systematic approach, based on performance modeling, is required for scalable software development for optimized molecular simulations on the future HPC architectures. Recently, we have ported and optimized LAMMPS (a popular molecular simulations code) on GPU-enabled Linux clusters, providing 10-20 fold speed-ups for biomolecular simulations. Our emphasis is on developing performance models that will allow optimal utilization of the heterogeneous computing resources for best scientific productivity. Codes including LAMMPS, AMBER and AutoDock are being optimized for heterogeneous architectures. Click here for details on NVIDIA's web-site.
Our GPU-LAMMPS is distributed under Open Source Software model, see our Software section for obtaining a copy. It is capable of running simulations based on CHARMM and AMBER force-fields and can be run with CHARMM/AMBER topology, coordinate files (after conversion with provided scripts).
Building upon the preliminary successes, we are using a systematic approach engineer molecular dynamics (MD) and molecular docking codes on the future heterogeneous architectures, including the Exascale. In particular, our focus includes:
- 1. Optimize and scale MD codes LAMMPS and AMBER, and docking code AutoDock on GPU-enabled HPC architectures by matching the software requirements with hardware. Performance models have been developed based on workload characterization. These performance models are being used for developing alternate data and communication patterns to exploit the computational power of multi-cores processors coupled with GPUs.
- 2. We are extending the developed code to other types of heterogeneous architectures including multi-core and many-core processors, ARM processors and field-programmable gate arrays (FPGAs). The performance models are being extended to other heterogeneous devices and will allow quantitative comparison of the performance improvements on alternate hardware devices. Further, the MD performance will be optimized based on an independent agent based framework to manage hardware and off-loaded computations.
- 3. We are also aiming to improving the end-user productivity by integrating fault-tolerance and application auto-turning strategies for optimal MD performance. Novel strategies for application health monitoring, fault detection and recovery are being inbuilt into the MD codes. An auto-tuning methodology that explores the highly multi-dimensional space of the hardware configuration parameters is being developed. This auto-tuning methodology will allow end-user to automatically achieve the optimal time-to-solution on heterogeneous architectures.
Application driven Fault-tolerance on Extreme-scale Architectures
Application resiliency will be one of the critical factors in determining scientific productivity on the upcoming High Performance Computing (HPC) architectures including the Exascale. Applications oblivious and incapable of handling transient soft and hard errors could waste computing resources, or worse, yield misleading scientific insights. To overcome this challenge, We are exploring a novel application driven silent error detection and recovery strategy based on application self-health monitoring. Our methodology is based on utilization of application output that follows known patterns as indicators of application's health and violation of these patterns could be indication of faults. Information from system monitors that report hardware and software health status is used to corroborate faults. Collectively this information is used by a fault coordinator agent to take preventive and corrective measures. This cooperative fault management system utilizes the Fault-Tolerance Backplane (FTB) as a communication channel.
The Fault Coordination Framework uses two critical components: the Application Monitor Agent and the Fault Coordinator Agent. The Application Monitor Agent interacts with the application to monitor any deviations in the simulations. The important features of our agent include: it does not require any code modifications to the application code, it runs independently of the application and and is completely re-usable with a variety of applications. The Fault Coordinator Agent is capable of utilizing information from system monitors that report hardware and software health status is used to corroborate the faults captured by the Application Monitor Agent.
This framework are has been successfully used with molecular dynamics simulations and quantum chemistry calculations on scalable clusters to handle memory and I/O corruptions.
Open Source Software for scientific computations
We are interested in developing Open Source Software for scientific applications. We have developed VigyaanCD, a Live Linux based bio/chemical software workbench that provides modeling tools for biomolecular and chemical modeling for education and research purposes. VigyaanCD has been downloaded over 250,000 times around the world.
Our GPU-LAMMPS for biomolecular simulations is also available under Open Source Software model, see our Software section for obtaining a copy. The software is capable of running simulations based on CHARMM and AMBER force-fields and can be run with CHARMM/AMBER topology, coordinate files (after conversion with provided scripts). It is capable of running on single workstations as well as scalable GPU-enabled Linux clusters.
An early release of the Application Monitor Agent is also available for testers. In combinations with the Fault-Tolerance Backplane (FTB), this agent can be used for application to self-monitor health.
Please see the Software section for downloading our software.